chemfp simhistogram¶
The “chemfp simhistogram” command-line tool generates a histogram of Tanimoto scores between all fingerprint pairs in one dataset (by convention this is called “the targets”) or between two datasets (by convention the “queries” and the “targets”).
These histograms can be used to assess the overall inter- or intra- similarity.
The histogram is distributed uniformly across --num-bin bins. By
default all bins except the last are closed/open, and the last bin is
closed/closed to include 1.0 For example, if there are 10 bins then
the first bin contains the counts of scores in the range [0.0, 0.1)
and the last contains the counts of scores in the range [0.9, 1.0].
Use --identity-bin so the counts with score 1.0 are placed into an
additional bin in the range [1.0, 1.0].
When comparing query and target fingerprints, use --include-inputs
to also compute the histogram of the queries with itself and the
targets with itself.
By default the histogram samples up to 25,000 distinct pairs to
generate the histogram. Use --num-samples to change the size or
--full to evaluate all pairs.
This command is also available as “chemfp simhist”.
The following image shows the distribution of Tanimoto scores between ChEBI and ChEMBL, and compares that distribution with the distribution of scores for each dataset with itself.
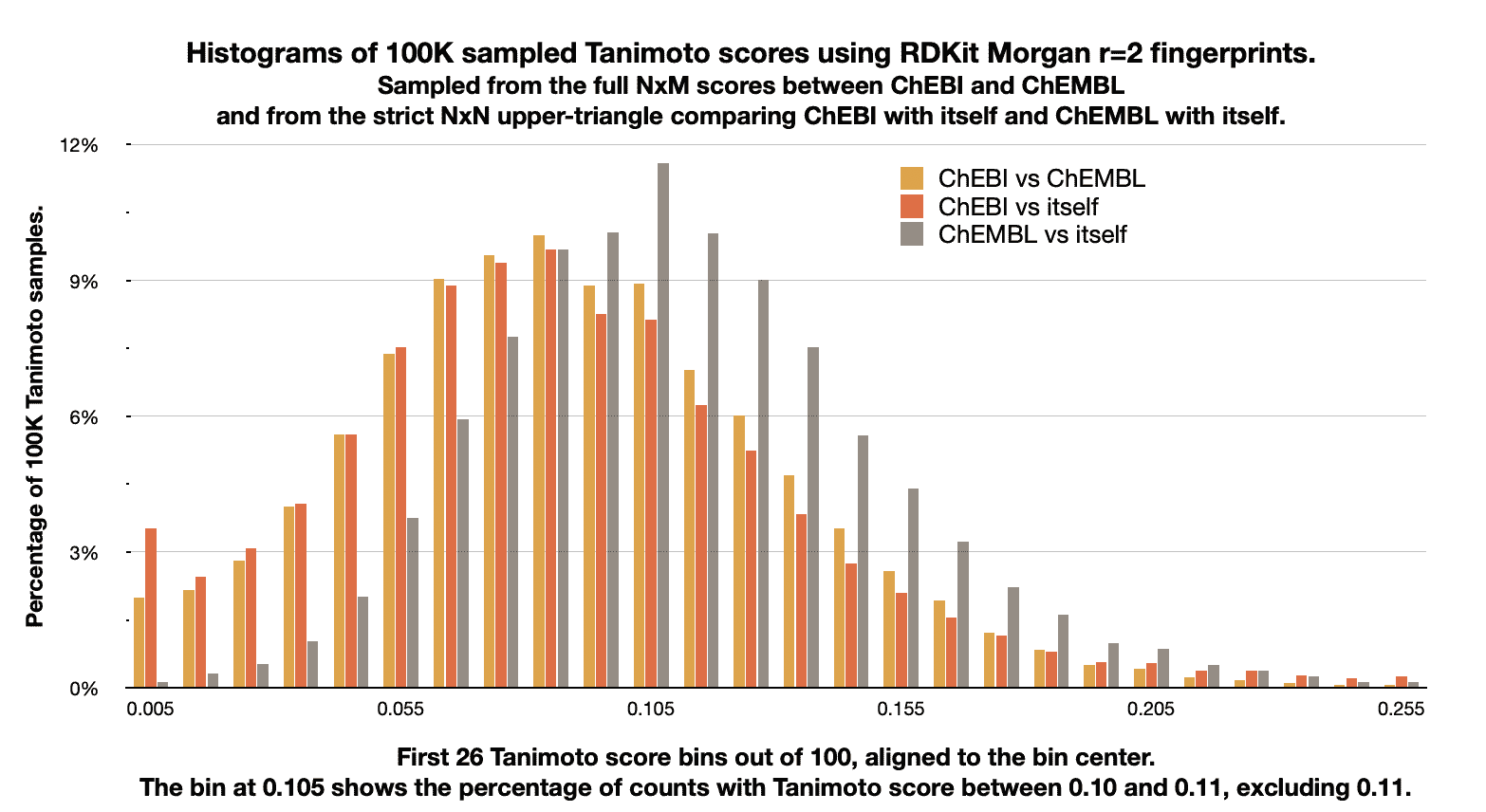
It was generated on the command-line with the following command-line,
the progress bar, and the first 27 lines of output (the first line of
output starts with #simhistogram/1):
% chemfp simhist --queries chebi_morgan.fpb chembl_35.fpb \
--num-samples 100_000 --include-inputs
NxM: 100%|██████████████████████████| 100000/100000 [00:03<00:00, 31624.51/s]
#simhistogram/1 identity-bin=0
#type=sample matrix-type=NxM bins=100 num-samples=100000 seed=1719484950
#size=281256950220 queries=113658 targets=2474590
#identical=0
#average=0.090 min=0.085 max=0.095
#queries-type=sample matrix-type=upper-triangular bins=100 num-samples=100000 seed=2822803993
#queries-size=6459013653 N=113658
#queries-identical=3
#queries-average=0.098 min=0.093 max=0.103
#targets-type=sample matrix-type=upper-triangular bins=100 num-samples=100000 seed=336081226
#targets-size=3061796596755 N=2474590
#targets-identical=0
#targets-average=0.111 min=0.106 max=0.116
#queries=chebi_morgan.fpb
#targets=chembl_35.fpb
start end count percent queries_count queries_percent targets_count targets_percent
0.00 0.01 1981 1.981 3492 3.492 110 0.110
0.01 0.02 2166 2.166 2436 2.436 278 0.278
0.02 0.03 2837 2.837 3087 3.087 533 0.533
0.03 0.04 4048 4.048 4014 4.014 1043 1.043
0.04 0.05 5646 5.646 5696 5.696 2023 2.023
0.05 0.06 7541 7.541 7633 7.633 3756 3.756
0.06 0.07 8779 8.779 8854 8.854 5775 5.775
0.07 0.08 9532 9.532 9401 9.401 7931 7.931
0.08 0.09 9937 9.937 9563 9.563 9842 9.842
0.09 0.10 8784 8.784 8215 8.215 9944 9.944
0.10 0.11 8901 8.901 8102 8.102 11587 11.587
The rest of this chapter contains the output from chemfp simhistogram --help.
chemfp simhistogram command-line options¶
The following comes from chemfp simhistogram --help:
Usage: chemfp simhistogram [OPTIONS] TARGETS
Generate a histogram from full or sampled Tanimoto scores.
Options:
-q, --queries PATH Filename containing the query fingerprints.
--in, --target-format FORMAT Input target format (default uses the file
extension, else 'fps')
--query-format FORMAT Input query format (default uses the file
extension, else 'fps')
--num-bins, --bins N Number of bins in the histogram (default:
100). [1<=x<=1000000]
--num-samples N Number of samples (-1 is the same as --full
search) (default: 25_000).
[-1<=x<=9223372036854775807]
--full Do a full search (ignore --num-samples).
--identity-bin / --no-identity-bin
With --identity-bin, place the 1.0 scores in
its own bin.
--include-inputs / --exclude-inputs
Use --exclude-inputs to omit the query and
target NxN columns in a NxM histogram.
--seed N Specify the random number generator seed
between 0 and 2**64-1, inclusive, or use -1
to have one picked at random (default: -1)
-o, --output FILENAME Output filename (default is stdout)
--out FORMAT Output format (default guesses from the
output filename, or is 'txt'
--include-metadata / --no-metadata
With --no-metadata, do not include header
metadata in 'txt' output format.
-j, --num-threads N The number of threads to use. If not
specified, 4 for sample, else -1.-1 means
the default value (which is 8 for this
computer), and can be set using
$OMP_NUM_THREADS. 0 and 1 both mean single-
threaded.
--no-mmap Don't use mmap to read uncompressed FPB
files. May give better performance on
networked file systems, at the expense of
higher memory use.
--times / --no-times Write timing information to stderr
--progress / --no-progress Use --no-progress to disable the default
progress bar.
--help Show this message and exit.
Generate a histogram of Tanimoto scores between pairs of fingerprints. The
pairs can be from the same dataset (NxN) or from two different datasets
(NxM).
By default, randomly sample 25,000 distinct pairs and generate 100 bins. If
there are fewer than 25,000 pairs then compute all available pairs. Use
`--num-samples` to change the sample size or use `--full` to always compute
all pairs. Use `--num-bins` to change the number of bins. Use `--seed` to
set RNG seed for random sampling.
By default (or with `--no-identity-bin`), all bins excepting the last are
half-closed, half-open intervals, that is, with B bins the first bin
contains the count of all scores 0 <= score < 1/B, the second bin the counts
of all scores 1/B <= score < 2/B, and so on. The B-th bin (the last bin) is
for the closed interval (B-1)/B <= score <= 1.0.
With `--identity-bin` then the B-th interval is half-closed, half-open and a
new identity bin is added of the count of scores = 1.0.
In NxN search only the strict upper triangle is used, that is, the diagonal
and lower triangle are ignored.
The following generates 10 bins based on 100,000 sampled pairs from ChEMBL
35 with itself:
% chemfp simhistogram chembl_34.fpb --num-bins 10 --num-samples 100000
#simhistogram/1 identity-bin=0
#type=sample matrix-type=upper-triangular bins=10 num-samples=100000 seed=4133835463
#size=3061796596755 N=2474590
#identical=0
#average=0.11 min=0.06 max=0.16
#targets=chembl_35.fpb
start end count percent
0.0 0.1 41239 41.239
0.1 0.2 56229 56.229
0.2 0.3 2438 2.438
0.3 0.4 83 0.083
0.4 0.5 6 0.006
0.5 0.6 5 0.005
0.6 0.7 0 0.000
0.7 0.8 0 0.000
0.8 0.9 0 0.000
0.9 1.0 0 0.000
The '#size' reports the number of pairs in the upper-triangle, which is over
3 trillion because ChEMBL 35 has about 2.47 million records. The
'#identical' reports there are no scores of 1.0. The average Tanimoto (based
on the histogram bins) is between 0.06 and 0.16, with the midpoint of 0.11.
The following uses the shorter alias 'simhist' to compare the query and
target fingerprint and output 5 bins partitioning the range 0.0 to 1.0, plus
the `--identity-bin` which adds the last bin for the number of 1.0 scores,
which is 1 in this case. The output is in csv format.
% chemfp simhist --queries queries.fps targets.fps --num-bins 5 \
--identity-bin --out csv
start,end,count,percent
0.0,0.2,6453,64.530
0.2,0.4,3354,33.540
0.4,0.6,180,1.800
0.6,0.8,9,0.090
0.8,1.0,3,0.030
1.0,1.0,1,0.010
For NxM search, use `--include-inputs` to also include the symmetric
histograms for the queries and targets, along with their respective metadata
fields.
% chemfp simhist --queries queries.fps targets.fps --num-bins 5 \
--identity-bin --include-inputs
#simhistogram/1 identity-bin=1
#type=full matrix-type=NxM bins=5
#size=10000 queries=100 targets=100
#identical=1
#average=0.18 min=0.08 max=0.28
#queries-type=full matrix-type=upper-triangular bins=5
#queries-size=4950 N=100
#queries-identical=6
#queries-average=0.25 min=0.15 max=0.35
#targets-type=full matrix-type=upper-triangular bins=5
#targets-size=4950 N=100
#targets-identical=41
#targets-average=0.25 min=0.15 max=0.35
#queries=queries.fps
#targets=targets.fps
start end count percent queries_count queries_percent targets_count targets_percent
0.0 0.2 6453 64.530 2219 44.828 2422 48.929
0.2 0.4 3354 33.540 2145 43.333 1994 40.283
0.4 0.6 180 1.800 412 8.323 156 3.152
0.6 0.8 9 0.090 74 1.495 171 3.455
0.8 1.0 3 0.030 94 1.899 166 3.354
1.0 1.0 1 0.010 6 0.121 41 0.828
For `--include-inputs` the `--seed` is used to seed Python's built-in RNG to
determine the query and target sampling seeds.