What’s new in chemfp 5.0¶
Version 5.0b2 released 20 August 2025.
The main additions to chemfp 5.0 are:
Update the FPB format to handle over 1 billion fingerprints.
New chemfp shardsearch command-line tool which does similarity search across multiple target files and merges the result.
New chemfp simhistogram / chemfp simhist command-line tool and corresponding
chemfp.simhistogram()high-level API function to create a histogram of similarity scores.Experimental support to generate sparse count fingerprints using RDKit along with methods to convert sparse count fingerprints to binary fingerprints and vice versa.
Implementations of the 4860-bit Klekota-Roth fingerprint for the OpenEye and RDKit toolkits.
See below for more details.
Backwards-incompatible changes¶
Dropped support for Python 3.8.
If no explicit fingerprint type is specified then rdkit2fps now generates Morgan fingerprints with radius 3 instead
of RDKit’s Daylight-like fingerprints. Use an explicit --RDK to
the command-line to get the RDKit fingerprint type. This change was
documented in the chemfp 4.2 release notes.
The toolkit helper functions matching the pattern “from_{format}”,
“to_{format}”, “from_{format}_file” and “to_{format}_file” (like
chemfp.rdkit_toolkit.from_smi to parse a SMILES record into a
molecule) were added in chemfp 4.0 and documented as deprecated in
chemfp 4.1. Using them in chemfp 4.2 generated a DeprecationWarnings.
With chemfp 5.0 they have been removed. Instead, use the
“parse_{format}” and “create_{format}” functions, like
parse_smi() instead of from_smi().
The chemfp.bitops functions byte_difference() and
hex_difference() have been removed. Using them generated a
PendingDeprecationWarning in chemfp 4.1 and a DeprecationWarning in
chemfp 4.2. Instead, use byte_xor
and hex_xor.
The fingerprint type method from_mol() and from_mols()
were documented as deprecated in chemfp 4.2. Using them now generates
a DeprecationWarning. They will be removed by chemfp 5.2. Instead, use
compute_fingerprint() or compute_fingerprints().
The high-level chemfp.simsearch(), chemfp.maxmin(),
chemfp.spherex() chemfp.heapsweep() functions provide
access to the low-level data structure as the attribute out.
Originally this was stored in the result attribute but experience
showed that referring to the low-level object as result.result was
more confusing than using result.out. The out alias for
result was added in chemp 4.2. Using result in chemfp 5.0
gives a PendingDeprecationWarning. In chemfp 5.1 it will give a
DeprecationWarning, with removal in chemfp 5.2.
WARNING: RDKit 2024.09.3 fixed a bug in how Morgan fingerprints
handled chiral atoms (see “Morgan fingerprints with chirality
distinguish chiral atoms too early”), causing some
fingerprints to change when includeChirality=1. This does not affect
the default Morgan fingerprints, which has includeChirality=0.
Previously this would trigger a version number change in the chemfp fingerprint, from RDKit-Morgan/2 to RDKit-Morgan/3. However, by design the version number change triggers downstream compatibility warnings, because chemfp has no way to figure out if the version difference is meaningful for a given set of options, and I haven’t figured out a good solution.
WARNING: RDKit versions 2024.9.2 changed the implementation of
includeRedundantEnvironments=1 but the RDKit-Morgan version number
has not changed, for the same reason as above.
NOT YET: The chemfp 4.2 release notes mention that chemfp 5.0 will change so that progress bars aren’t shown unless there is a certain minimum delay. This change has not been done, but may in the future.
NOT YET: The chemfp 4.2 release notes mention that the “npz” JSON metadata array format “will likely change”, to be more consistent with then-new simarray JSON. This has not yet happened but may in the future.
Updated FPB format¶
The original FPB format could not handle more than about 270 million fingerprints due to an implementation detail in the “HASH” chunk. Chemfp 5.0 supports a new “HSH2” chunk which should handle at least 2 billion fingerprints, though it has only been tested with 1 billion fingerprints.
See the FPB specification for details.
Warning
Even though the --max-spool-size of the
fpcat command-line tool helps limit the amount
of RAM needed, the final FPB generation step loads all of the ids
into memory. You will likely need about 35-40 GB of RAM to
generate an FPB file with 1 billion fingerprints.
simhistogram¶
The new “simhistogram” feature computes a histogram of the Tanimoto
similarity scores in a fingerprint dataset with itself (“NxN”), or
between two different fingerprint datasets (“NxM”). It is available
using the chemfp simhistogram
command-line tool or in the Python API using the
chemfp.simhistogram() function.
The command-line tool, when used to generate an NxM histogram, can also include the NxN histograms for each of the two input dataset.
These histograms can give a sense of the overall similarity distribution. For example, the following image shows the distribution of Tanimoto scores between ChEBI and ChEMBL, and compares that distribution with the distribution of scores for each dataset with itself.
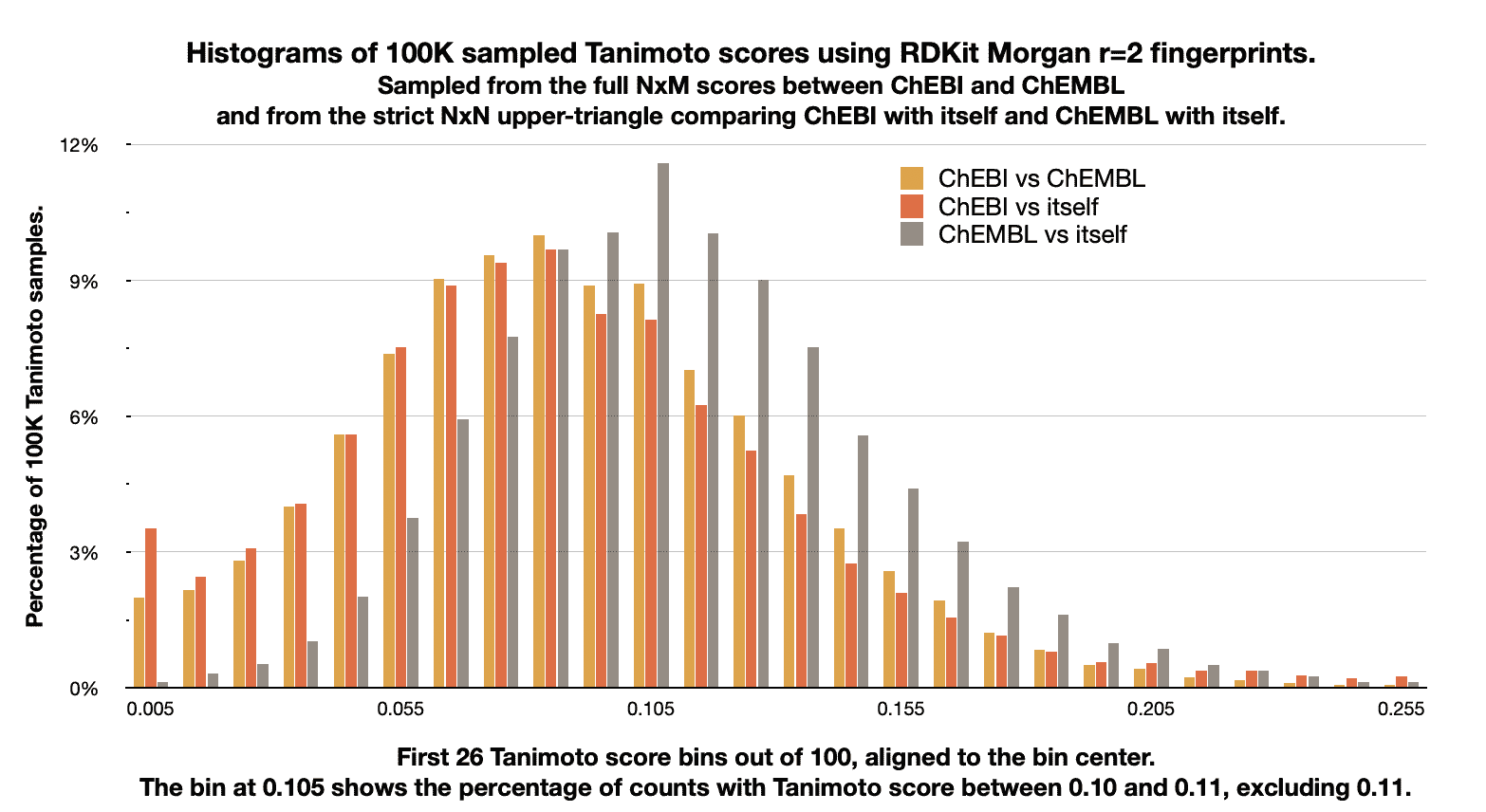
It was generated on the command-line with the following command-line,
the progress bar, and the first 27 lines of output (the first line of
output starts with #simhistogram/1):
% chemfp simhist --queries chebi_morgan.fpb chembl_35.fpb \
--num-samples 100_000 --include-inputs
NxM: 100%|██████████████████████████| 100000/100000 [00:03<00:00, 31624.51/s]
#simhistogram/1 identity-bin=0
#type=sample matrix-type=NxM bins=100 num-samples=100000 seed=1719484950
#size=281256950220 queries=113658 targets=2474590
#identical=0
#average=0.090 min=0.085 max=0.095
#queries-type=sample matrix-type=upper-triangular bins=100 num-samples=100000 seed=2822803993
#queries-size=6459013653 N=113658
#queries-identical=3
#queries-average=0.098 min=0.093 max=0.103
#targets-type=sample matrix-type=upper-triangular bins=100 num-samples=100000 seed=336081226
#targets-size=3061796596755 N=2474590
#targets-identical=0
#targets-average=0.111 min=0.106 max=0.116
#queries=chebi_morgan.fpb
#targets=chembl_35.fpb
start end count percent queries_count queries_percent targets_count targets_percent
0.00 0.01 1981 1.981 3492 3.492 110 0.110
0.01 0.02 2166 2.166 2436 2.436 278 0.278
0.02 0.03 2837 2.837 3087 3.087 533 0.533
0.03 0.04 4048 4.048 4014 4.014 1043 1.043
0.04 0.05 5646 5.646 5696 5.696 2023 2.023
0.05 0.06 7541 7.541 7633 7.633 3756 3.756
0.06 0.07 8779 8.779 8854 8.854 5775 5.775
0.07 0.08 9532 9.532 9401 9.401 7931 7.931
0.08 0.09 9937 9.937 9563 9.563 9842 9.842
0.09 0.10 8784 8.784 8215 8.215 9944 9.944
0.10 0.11 8901 8.901 8102 8.102 11587 11.587
The simhist subcommand is an alias for simhistogram.
This shows the default output format, which includes a header second
containing metadata about the calculation. Use --out or an
appropriate output filename extension to get just the histogram data
formatted as “tsv” or “csv”.
simhistogram output header¶
The output contains a header followed by the histogram lines. The first header line identifies the format version and subtype. The #type line describes how the histogram was generated, including any parameters. In this case it contains 100,000 scores randomly sampled from an NxM similarity matrix, placed into 100 bins. The initial RNG seed, in this case generated by Python’s built-in RNG, is 1719484950.
The #size line reports the ChEBI and ChEMBL datasets have 113,658 and 2,474,590 fingerprints, respectively, so the full NxM comparison matrix contains over 281 billion scores.
Based on the histogram data (computing by the weighted average of
bin count * bin center), the #average ChEBI vs. ChEMBL score is
0.090. This may not be the actual average score as the actual scores
may be anywhere in the bin, but it’s certainly between 0.093 and
0.103, because in this case the bin width is 0.1 and there are no
#identical scores. (Internally chemfp always places the identical
scores in their own bin, with a bin center of 1.0 and width of 0.0,
even with the default of --no-identity-bin, which affects how
average, min, and max are calculated.)
The lines starting #queries- give the corresponding details for
strictly upper-diagonal triange of the similarity scores of the
--queries dataset with itself, and the lines starting #targets-
do the same for the targets.
The #queries and #targets lines describe the source for the queries and targets, which is typically the filename.
By default (with --no-identity_bin) there are N bins and each bin
has the width N / --num-bins. The first N-1 bins are
half-closed/half-open, while the last bin is closed. For example, if
there are 2 bins then bin 0 contains the count of scores S where 0
<= S < 0.5, and bin 1 contains the count of scores where 0 <= S <=
0.5.
With --identity-bin, the first N bins are all half-closed/half
open, and last bin contains the count of scores which are exactly 1.0.
simhistogram output data¶
The lines after the header contain the histogram data, starting with column titles.
If the output is a single histogram (the default) then the column
titles are “start”, “end”, “count”, and “percent”. If the output
contains three histograms (using --include-inputs) then the
column titles are “start”, “end”, “count”, “percent”, “queries_count”,
“queries_percent”, “targets_count”, and “targets_percent”.
The “start” and “end” columns contain the bin start and end positions,
which for bin i<=N are i/N and (i+1)/N, respectively. The
optional --identity- bin start and end positions are always 1.0.
The “count” column contains the number of scores in the given bin, and “percent” is the number of scores scaled by the number of samples.
The “queries_count”, “queries_percent”, “targets_count”, and “targets_percent” columns contain the query-specific or target-specific equivalents to “count” and “percent”.
The “csv” or “tsv” output formats only show the output data. For example, the following generates a histogram in CSV format with 11 bins (10 bins plus a special bin for scores which are exactly 1.0) using 100 million samples, without duplicates, of the over 3 trillion Tanimoto scores when comparing ChEMBL 35 with itself:
% chemfp simhist chembl_35.fpb --num-bins 10 --identity-bin
--num-samples 100_000_000 --seed 12345 --out csv --no-progress
0.0,0.1,41311159,41.311
0.1,0.2,56230689,56.231
0.2,0.3,2362965,2.363
0.3,0.4,81778,0.082
0.4,0.5,9764,0.010
0.5,0.6,2465,0.002
0.6,0.7,776,0.001
0.7,0.8,267,0.000
0.8,0.9,93,0.000
0.9,1.0,29,0.000
1.0,1.0,15,0.000
The --seed initializes the RNG used for sampling, making the
output reproducible. The --no-progress disables the progress
bar.
High-level chemfp.simhistogram function¶
Use the new chemfp.simhistogram() function to create a
histogram using Python.
The API is influenced by the NumPy histogram
function API, which uses bins (NOT num_bins!) to specify the
number of bins. The histogram result can be assigned to (hist,
bin_edges) containing the histogram values and the bin edgets,
respectively.
For example:
>>> import chemfp
>>> hist, bin_edges = chemfp.simhistogram(targets="chembl_35.fpb", bins=5, progress=False)
>>> hist
array('Q', [24369, 627, 4, 0, 0])
>>> bin_edges
array('d', [0.0, 0.2, 0.4, 0.6, 0.8, 1.0])
The actual SimHistogramResult object (which implements the
item lookup [0] and [1] used for the above 2-element tuple assignment)
stores more information about the results, query parameters, and timings:
>>> result = chemfp.simhistogram(targets="chembl_35.fpb", bins=10,
... num_samples=100_000_000, seed=12345)
scores: 100%|█████████████████████████████████████| 100M/100M [00:19<00:00, 5.12M/s]
>>> result
SimHistogramResult(<sampled upper-triangle, 100000000 pairs (0.003%), seed=12345,
10 bins>, times=<total: 19.56 s>)
>>> result.bins
array('Q', [41311159, 56230689, 2362965, 81778, 9764, 2465, 776, 267, 93, 44])
>>> result.edges
array('d', [0.0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0])
>>> result.num_identical
15
>>> result.seed
12345
>>> f"{result.num_processed:,} of {result.total_size:,}"
'100,000,000 of 3,061,796,596,755'
>>> result.times
{'load_queries': None, 'load_targets': 0.00724029541015625, 'init': 0.0054018497467041016,
'process': 19.547301769256592, 'total': 19.55998182296753}
>>> result.close()
sparse count fingerprints¶
Chemfp 5.0 adds experimental and limited support for sparse count fingerprints.
The new FPC format is a text-based format for sparse count fingerprints.
The new chemfp rdkit2fpc subcommand uses RDKit to generate sparse count fingerprints in FPC format from structure files.
The new chemfp fpc2fps subcommand implements several different methods to convert a sparse count fingerprint into a (dense) binary fingerprint in FPS or FPB format.
The new chemfp fps2fpc subcommand converts binary fingerprints into FPC format.
FPC format¶
The new FPC format for sparse count fingerprints is almost identical to the FPS format format for binary fingerprints. Here is an example:
#FPC1
#type=RDKit-MorganCount/2 radius=3 useFeatures=0
#software=RDKit/2024.09.5 chemfp/5.0
#date=2025-08-15T11:17:47+00:00
847433064,2245897107,2551683561 CHEMBL183419
864674487,1215180924,1600340860:2,2215059400,2245384272:2,2246728737:2,3542456614:2,3994088662:2 CHEMBL16264
The main difference is in the fingerprint records lines, where the hex-encoded fingerprint is replaced with a text encoding for sparse count fingerprints.
Each sparse count fingerprint contains a sequence of zero or more
features. If there are no features then the corresponding text
encoding is *, otherwise it’s a comma-separated list of
text-encoded features.
Each feature contains a feature id (also called a bit position), which is a non-negative integer less than 2^64, and a feature count, which is a non-negative integer less than 2^32.
The feature encoding is the string with the feature id, followed by a colon, followed by the count, as in “{feature id}:{feature count}”. For example, if the feature id is 87501 and the feature count is 505 then the encoded feature is “87501:505”.
As a special case, if the feature count is 1 then the feature may be encoded as just the feature id, eg, “87501” instead of “87501:1”.
A feature count of 0 is valid, in which case the feature may be ignored.
Lastly, the feature ids must be in increasing order.
rdkit2fpc¶
The rdkit2fpc subcommand uses RDKit to generate Morgan, RDKit (Daylight-like), AtomPair, and Torsion count fingerprints, with Morgan fingerprints of radius 3 as the default if a specific type is not given. Here are examples of each type using CO2 read from stdin as a SMILES file:
% echo "O=C=O carbon dioxide" | chemfp rdkit2fpc --morgan
#FPC1
#type=RDKit-MorganCount/2 radius=3 useFeatures=0
#software=RDKit/2024.09.5 chemfp/5.0
#date=2025-08-18T12:30:31+00:00
864942730:2,1517923320:2,2245900962,2962078081 carbon dioxide
%
% echo "O=C=O carbon dioxide" | chemfp rdkit2fpc --RDK
#FPC1
#type=RDKit-CountFingerprint/3 minPath=1 maxPath=7
#software=RDKit/2024.09.5 chemfp/5.0
#date=2025-08-18T12:30:49+00:00
1488142319,1628256115:2,1638855635,2172716083:2 carbon dioxide
%
% echo "O=C=O carbon dioxide" | chemfp rdkit2fpc --pair
#FPC1
#type=RDKit-AtomPairCount/3 minDistance=1 maxDistance=30
#software=RDKit/2024.09.5 chemfp/5.0
#date=2025-08-18T12:31:02+00:00
1721921:2,1723682 carbon dioxide
%
% echo "O=C=O carbon dioxide" | chemfp rdkit2fpc --torsion
#FPC1
#type=RDKit-TorsionCount/4 torsionAtomCount=4
#software=RDKit/2024.09.5 chemfp/5.0
#date=2025-08-18T12:31:05+00:00
* carbon dioxide
fpc2fps¶
Chemfp does not currently support similarity search directly on FPC files. Instead, sparse count fingerprints must be converted to FPS or FPB format using the chemfp fpc2fps command, then searched as binary fingerprints.
The FPC format supports (2^64 * 2^32) = 2^96 distinct features. There is no general purpose method to convert them to a short dense fingerprint (typically with a few thousand bits) where the generalized Jaccard index/Tanimoto score between two fingerprint pairs is preserved.
On the other hand, approximate methods may be good enough. Let N
be the number of bits in the binary fingerprint and for a given
feature let i be the feature id (ie, the sparse bit position) and
n be the feature count.
One classic method is to “fold” each feature. For each feature,
compute the binary bit position b by computing b = i % N then
set that bit to 1. Ignore the count.
This is the default RDKit method, following the footsteps of Daylight which introduced this approach.
The “fold” method only distinguishes between counts of zero and not-zero, and cannot distinguish between two features ids which fold to the same bit. In the following, which uses the Morgan3 count fingerprint for CO2, you can see that four output bits are set. They come from the four non-zero feature ids:
% printf '30:2,1517923320:2,2245900962,2962078081\tCO2\n' | \
chemfp fpc2fps --num-bits 32 --fold
#FPS1
#num_bits=32
#type=fold/1 num_bits=32
06000041 CO2
The fpc2fps default method is “superimpose”, which applies aspects of the Zatocoding method of Calvin Mooers and the RNG-seed of Daylight fingerprints to better emulate count fingerprints
The key observation is that many binary fingerprints, like the folded circular Morgan fingerprints, set fewer than 5% of the bits, and the total count (the sum of count for each feature) for the original sparse count fingerprints is still small relative to the binary fingerprint size.
For example, of the 2.37 million Morgan fingerprints with radius=3 computed from ChEMBL 33, only 62,000 have a total count of 100 or more, and only 600 have a total count of 200 or more, with CHEMBL3298848 having the highest total count at 284.
On the other hand, the corresponding binary fingerprints are typically 1024 to 4096 bits. These many zeros could be used to encode count information.
The “superimpose” method can be described with the following pseudocode:
binary_fp= BinaryFingerprint(num_bits)
for (feature_id, feature_count in count_fp.features:
rng = RNG(feature_id)
feature_count = min(feature_count, max_feature_count)
for _ in range(feature_count * bits_per_count):
bitno = rng.randrange(num_bits)
binary_fp.SetBitToOn(bitno)
That sets count * bits_per_count output bits for each feature id,
selected at random (with replacement) from an RNG seeded by the
feature id, with a possible upper limit to the count. As
bits_per_count and num_bits increase, the effect of collisions
decreases, though no one has yet characterized its effectiveness.
In the following you can see that five output bits are set, while the
total feature count is 2+2+1+1= 6:
% printf '30:2,1517923320:2,2245900962,2962078081\tCO2\n' | \
chemfp fpc2fps --num-bits 32 --superimpose
#FPS1
#num_bits=32
#type=superimpose/1 num_bits=32
04010045 CO2
The “rdkit” method implements RDKit’s “count simulation” option, which
takes a C count bounds {c_j} where 0 <= j < C, to first create
a dense count fingerprint with N / C counts by merging the sparse
count fingerprint into the dense one with the sum S of all feature
counts where i % (N/C) is the same, then using the sum count S
at position i % (N/C) to set i % (N/C) * C + j to 1 iff S >=
c_j.
If you can figure out what I wrote here, well done! If not, see chemfp fpc2fps –help-methods for more details.
In the following you can see that size output bits are set, which
corresponds to the total feature count of 2+2+1+1=6:
% printf '30:2,1517923320:2,2245900962,2962078081\tCO2\n' | \
chemfp fpc2fps --num-bits 32 --rdkit --countBounds 1,2
#FPS1
#num_bits=32
#type=rdkit-count-sim/1 num_bits=32 countBounds=1,2
14000330 CO2
The “seq” method is for dense count fingerprints (that is, count
fingerprints with at most a few hundred feature ids F, in the
sequence 0, 1, ... F-1). The driving use case for “seq” encoding
is a fingerprint type defined by a few dozen SMARTS patterns, where
the count is the number of unique matches, and where the count is
typically small (eg, under 20) and a where a relatively small upper
bound (eg, 100) can be applied to each count.
These constraints mean the dense count fingerprint can be represented exactly as a binary fingerprint using unary encoding, which in turn means the generalized Jaccard index between two count fingerprints is identical to the Tanimoto similarity between two such binary fingerprints.
“Unary” encoding means that 1 distinct bit is set for each count, up
to a maximum number W. For example, if W is 5 then the
possible values, represented in binary, are “00000” for 0, “00001” for
1, “00011” for 2, “00111” for 3, “01111” for 4, and “11111” for 5 or
more.
In the following, the first bin is 8 bits/1 byte long and contains the value 4, which is “00001111” or “0f” in hex, the second bin is 8 bits/1 byte long and contains the value 0 or “00” in hex, the third bin is 16 bits/2 bytes long and contains the value 3, or “00000000 00000111”, which is “0700” in chemfp’s hex fingerprint representation, the fourth bin is 16 bits/2 bytes long and contains the value 9 or “00000001 11111111”, which is “ff01”, while the remaining values are 0:
% printf '0:4,2:3,3:7\tABC\n' | \
chemfp fpc2fps --seq --sizes 8,8,16,16,8,8
#FPS1
#num_bits=64
#type=seq/1 num_bits=64 sizes=8,8,16,16,8,8
0f000700ff010000 ABC
The other two methods are “scaled” and “seq-scaled” which are like “superimpose” and “seq” except they support a conversion scale from the feature count to the actual count to use. This can be used to create a hybrid between the “superimpose” and “rdkit” methods or to approximate TF-IDF (Term Frequency-Inverse Document Frequency).
Use chemfp fpc2fps –help-methods for more details about these methods and their respective command-line options. Please contact me if you are researching alternative methods of count emulation, which I think is good research topic.
fps2fpc¶
The chemfp fps2fpc subcommand converts binary fingerprints from an FPS or FPB file into a count fingerprint in FPC format. Given a binary fingerprint, the corresponding feature fingerprint is * if the fingerprint is empty, or a comma-separated list of the on-bit indices, in increasing order.
% printf '04010045\tXYZ\n' | chemfp fps2fpc
#FPC1
#type=fps2fpc/1
2,8,24,26,30 XYZ
Klekota-Roth fingerprints¶
The chemfp 5.0 release includes fast implementations of the 4860-bit
Klekota-Roth substructure fingerprint for the OpenEye and RDKit
toolkits, available in oe2fps and rdkit2fps using --KlekotaRoth. The corresponding chemfp
fingerprint types are “KlekotaRoth-OpenEye/1” and
“KlekotaRoth-RDKit/1”.
Klekota and Roth in “Chemical substructures that enrich for biological activity”, Bioinformatics, Volume 24, Issue 21, November 2008, Pages 2518–2525, doi:10.1093/bioinformatics/btn479 used a decision tree to identify privileged substructures associated with biological activity. The paper lists these substructures as 4860 SMARTS patterns.
I read the paper and thought the approach was both novel and interesting , with plenty of opportunity for someone to revist it with new datasets. However, very few papers cite it, and the only implementation I could find was for the CDK.
In asking around, I learned that one stumbing block was its poor performance. A direct implementation which does 4860 SMARTS substructure tests per molecule can process about 47 molecules per second using RDKit, or about 6 hours per million molecules.
By comparison, rdkit2fps takes about 5.5 minutes to generate 1 million Morgan3 fingerprints, and 17 minutes to generate the 1 million 166 key MACCS fingerprints.
I developed a method which analyzes the 4860 fingerprints to find dependencies between them. For example, if the pattern 4842 (”S”) does not match, then there is no reason to test for pattern 1148 “[!#1][SH]”). The method includes a few additional fragments, like nn, which were found to improve the overall performance. It then genenerates a Python module which implements an optimized fingerprint generator.
The faster implementation for RDKit takes about 21.5 minutes to process a million fingerprints, which is about 17 times faster than the direct implementation, and only about 25% slower than the MACCS keys.
Note
If you develop fingerprints based on a large number of SMARTS strings and want to improve generation performance then contact me to see if my method is applicable.
New APIs¶
Chemfp 5.0 added several new API functions.
The function chemfp.get_metadata_from_source() reads the
Metadata from a given source, which must be an FPS or FPB
file (FPC is not currently supported). This method can read the
metadata from a compressed FPB file without first decompressing the
entire file.
>>> import chemfp
>>> chemfp.get_metadata_from_source("chembl_34.fpb.zst")
Metadata(num_bits=2048, num_bytes=256, type='RDKit-Morgan/1 radius=2
fpSize=2048 useFeatures=0 useChirality=0 useBondTypes=1',
aromaticity=None, sources=[], software='RDKit/2022.09.4', date=None)
The FingerprintArena.close() method now explicitly discards its
reference to the internal fingerprint data storage, even for in-memory
arenas. This makes the memory use lifetime more explicit, which was
needed in shardsearch to ensure a large dataset was deallocated before
loading a new large dataset. The areana context manager will close
when exiting.
The new FingerprintArena.get_popcount_counts() method returns
an array of length num_bits + 1 where element i contains the
number of fingerprints with the popcount i. If the arena contains a
popcount index (which requires the fingerprints to be in popcount
order) then the array is computed directly from the index. If there is
no popcount index then by default it computes the popcount for each
fingerprint. If num_samples is an integer (and not None) then the
counts come from sampling num_samples distinct fingerprints and
computing their popcounts. Use seed to seed the underlying RNG.
>>> import chemfp
>>> arena = chemfp.load_fingerprints("maccs_50k1.fpb")
>>> len(arena)
50001
>>> arena.get_popcount_counts()
array('I', [0, 1, 0, 3, 6, 8, 12, 20, 9, 24, 24, 31, 33, 33, 48, 67,
62, 93, 92, 143, 164, 193, 247, 284, 328, 404, 441, 496, 534, 528,
583, 684, 704, 864, 845, 958, 1007, 1009, 1082, 1128, 1152, 1263,
1272, 1219, 1310, 1337, 1297, 1378, 1331, 1316, 1302, 1383, 1232,
1315, 1257, 1242, 1190, 1135, 1101, 1106, 1040, 961, 964, 940, 824,
759, 705, 642, 556, 534, 473, 409, 374, 324, 284, 250, 239, 216,
162, 191, 145, 112, 113, 81, 68, 74, 58, 47, 25, 20, 20, 20, 9, 8,
2, 4, 7, 6, 0, 3, 0, 0, 1, 0, 0, 0, 1, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0,
0, 0, 0, 0])
>>> sum(arena.get_popcount_counts())
50001
The new SearchResult.get_indices_ids_and_scores() method
returns a list of 3-element tuples containing the (index, id, score)
for each hit, in the current order.
>>> import chemfp
>>> arena = chemfp.load_fingerprints("chembl_34.fpb")
>>> query_fp = arena.fingerprints[1_000_000]
>>> result = arena.knearest_tanimoto_search_fp(query_fp, k=4)
>>> result.get_indices_ids_and_scores()
[(1000000, 'CHEMBL4456699', 1.0),
(923024, 'CHEMBL4462517', 0.631578947368421),
(854991, 'CHEMBL4564372', 0.6140350877192983),
(856777, 'CHEMBL4590721', 0.5081967213114754)]
The FingerprintType.get_type() method has a new complete
parameter. By default get_type returns the canonical string for the
fingerprint type, which might exclude some settings with default
parameters. (This generally occurs for newly added parameters, to
preserve compatibility with the older canonical string.) If complete
is True then the method returns the full type string:
>>> import chemfp
>>> chemfp.rdkit.atom_pair.get_type()
'RDKit-AtomPair/3 fpSize=2048 minDistance=1 maxDistance=30
countSimulation=1'
>>> chemfp.rdkit.atom_pair.get_type(complete=True)
'RDKit-AtomPair/3 fpSize=2048 minDistance=1 maxDistance=30
countSimulation=1 includeChirality=0 use2D=1 fromAtoms=None'
CDK¶
Chemfp 5.0 added support for new features in CDK 2.10 and 2.0.
Added support for the square planar, octahedral, and trigonal bipyramidal SmiFlavor flags added in CDK 2.10. They are ignored when using older versions of CDK.
Added version “2.10” Daylight, GraphOnly, and Hybridization fingerprinters. They support the “hashExplicitHydrogens” option, with a default of 0. (Explicit hydrogens are ignored.) The older version “2.0” versions always included explicit hydrogens.
Added version “2.10” ExtendedFingerprinter. Internally it uses a DaylightFingerprinter with hashExplicitHydrogens=0. The CDK API does not have a way to configure this value.
Added a “prepare” option to the CDK SMILES, SDF, molfile, and InChI readers. If true (the default) it identifies rings and perceives aromaticity.
Other¶
Chemfp 5.0 added support for Python 3.13, NumPy 2.0+, and click 8.2.
There are also some performance improvements with FPS parsing and FPB generation.
FIX: FPB generation can build larger-than-RAM datasets by saving partial data to temporary files (a “spool”) then merging the results. The estimator for the spool size was incorrect, which would limit spool sizes to under 1 GB. The estimator has been fixed.
Added specialized byte Tanimoto implementations for different fingerprint sizes. These is an internal detail which improves simhistogram performance, but does not affect the API. Use “report-byte-tanimoto” option to enable/disable selection reporting of the chosen implementation:
>>> import chemfp
>>> arena = chemfp.load_fingerprints("chembl_34.fpb")
>>> chemfp.simhistogram(targets=arena, progress=False)
SimHistogramResult(<sampled upper-triangle, 25000 pairs (0.0000%),
seed=1375723649, 100 bins>, times=<total: 2.37 s>)
>>> from chemfp import bitops
>>> bitops.set_option("report-byte-tanimoto", 0)
>>> chemfp.simhistogram(targets=arena, progress=False)
Byte tanimoto method: size256 (popcnt_128_128) num_bits: 2048
arena1: 0x10b5d9100 (128 byte aligned) storage_len1: 256
arena2: 0x10b5d9100 (128 byte aligned) storage_len2: 256
SimHistogramResult(<sampled upper-triangle, 25000 pairs (0.0000%),
seed=3331938674, 100 bins>, times=<total: 657.78 ms>)
FIX: Fixed a progress bar bug when the input is an FPB file. Thanks to Pavel Polishchuk who reported this bug.